Adrenogenitales Syndrom
Das Adrenogenitale Syndrom (AGS) ist eine angeborene Störung der Hormonbildung der Nebennierenrinde. Die Nebennierenrinde bildet das Streßhormon Cortisol, welches für die Widerstandsfähigkeit gegenüber Streß und die Aufrechterhaltung des Kreislaufes und Blutzuckers verantwortlich ist. Gleichzeitig wird das Hormon Aldosteron für die Aufrechterhaltung des Salzhaushaltes gebildet. Als weitere Hormone kann die Nebennierenrinde männliche Geschlechtshormone (sogenannte Androgene) produzieren. Beim Adrenogenitalen Syndrom kommt es durch eine angeborene, genetische Veränderung im Syntheseweg zur verminderten Bildung von Cortisol und Aldosteron sowie zu einer vermehrten Bildung männlicher Hormone (siehe Abbildung und Tabelle). Der Begriff Adrenogenitales Syndrom beschreibt mehrere Erkrankungen, die nach dem genetisch veränderten Eiweiß (Enzym) benannt werden. Die häufigste Form ist der 21-Hydroxylasemangel.
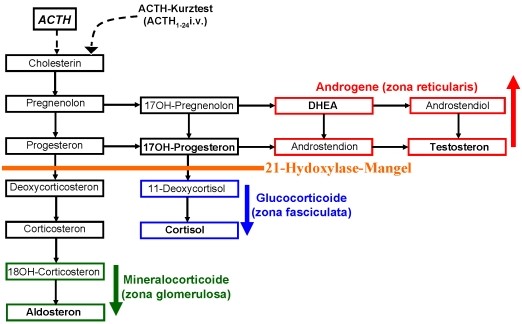
Abbildung: Störungen der Hormonbildung der Nebenniere beim AGS (21-Hydroxylase-Mangel). Wichtigstes Androgen ist das DHEA, wichtigstes Glucocorticoid das Cortisol, wichtigstes Mineralocorticoid das Aldosteron.

Tabelle: Zonen der Nebennierenrinde mit hormonellen Dysfunktionen bei AGS
Häufigkeit
Die Häufigkeit des klassischen 21-Hydroxylasemangels liegt bei ca. 1 Patienten auf 13.000 Einwohner. Die Frequenz der milder ausgeprägten "nicht-klassischen" Fälle ist nicht genau bekannt. Wahrscheinlich ist sie jedoch höher als die der klassischen AGS-Formen.
Beschwerden
Das klassische AGS ist charakterisiert durch eine vorgeburtliche Vermännlichung des äußeren Genitales bei Mädchen. Es gibt Formen mit und ohne einen Salzverlust, der zu einer Gedeihstörung aber auch Schock und Koma im Säuglingsalter führen kann. Jungen fallen als Neugeborene einzig durch den Salzverlust auf. Bei beiden Geschlechtern kommt es jedoch im weiteren Kindesalter durch die vermehrte Bildung von männlichen Hormonen zu Hochwuchs, Akne, vorzeitiger Genitalbehaarung, Stimmbruch, ausbleibender Regelblutung etc. Langfristig bleiben unbehandelte Kinder zu klein. Im Erwachsenenalter sind Patienten häufig von Übergewicht, Stoffwechselveränderungen und Unfruchtbarkeit betroffen. Weiter besteht ein lebenslanges Risiko für ein krisenhaftes Entgleisen der Erkrankung, wenn die Therapie nicht richtig durchgeführt wird.
Beim nicht klassischen oder late-onset AGS kommt es zu keiner vorgeburtlichen Vermännlichung. Die Ursache ist ein milderer Enzymdefekt, der bis in das Schulalter oder das junge Erwachsenenalter ohne Symptome bleiben kann. Zeichen können eine vorzeitige Schambehaarung, Akne und ein relativer Hochwuchs sein. Manche Patientinnen fallen erst durch eine ausbleibende Regelblutung oder einen unerfüllten Kinderwunsch auf.
Diagnostik
Im Neugeborenenscreening werden alle Kinder auf den 21-Hydroxylasemangel untersucht. Man entdeckt jedoch nur die klassischen Formen. Die Diagnose kann ansonsten anhand einer Blutentnahme durch die Bestimmung der Nebennierenhormone und männlichen Hormone erfolgen. Manchmal kann ein nicht klassisches AGS nur durch einen sogenannten ACTH Stimulationstest nachgewiesen werden. Hierbei wird die Nebenniere durch Gabe der im Körper vorhandenen Substanz ACTH zur Ausschüttung von Hormonen angeregt. Die Sicherung der Diagnose AGS erfolgt durch eine genetische Untersuchung einer Blutprobe.
Therapie
Ziele der AGS-Therapie sind der Ersatz der Glucocorticoide (Cortisol) und der Mineralocorticoide (Aldosteron) und die Normalisierung der männlichen Hormone (Androgene) (Tabelle). Die Therapie des klassischen AGS besteht aus einer lebenslangen Medikation.
Ersatztherapie eines Glucocorticoidmangels: Hierzu benutzt man meistens das physiologische Hydrocortison (=Cortisol) und teilt zur Nachahmung der zirkadianen Rhythmik in 2-3 Tagesdosen (z.B. 75-25-0% oder 50-25-25%). Bei erhöhtem Streß wie z.B. bei fieberhaften Erkrankungen muss die Tagesdosis verdoppelt oder verdreifacht werden. Bei operativen Eingriffen muss zur Vermeidung von lebensbedrohlichen Krisen eine noch höhere Dosis gegeben werden. Jeder Patient muß daher einen Glucocorticoid-Notfallausweis besitzen.
Ersatztherapie eines Mineralocorticoidmangels: Die Mineralocorticoidsubstitution erfolgt durch die orale Gabe 9a-Fluoro-Hydrocortison (Fludrocortison = Astonin H®) pro Tag. Zur Kontrolle der richtigen Dosierung misst man Kalium, Natrium und den Blutdruck und stellt die Plasma-Renin-Konzentration (Laborwert) in den oberen Normbereich ein. Normalisierung der adrenalen Androgenüberproduktion: Aufgrund der im Rahmen des Enzymdefektes auftretenden Glucocorticoidmangels ist das ACTH gegenregulatorisch erhöht (Abbildung), was zur vermehrten adrenalen Androgensynthese mit den entsprechenden Symptomen (Tabelle) führt. Die physiologische Glucocorticoidgabe reicht meistens zur Suppression des ACTHs und der adrenalen Androgene nicht aus, so dass man im Erwachsenenalter häufiger mit länger wirkenden synthetischen Glucocorticoiden (Prednisolon oder Dexamethason) und Einnahme entgegen der zirkadianen Rhythmik arbeitet. Dosierungen oberhalb der Cushing-Schwelle (Prednisolon 5 mg, Dexamethason 0,5 mg) müssen aber unbedingt vermieden werden. Eine gute Suppressionstherapie verhindert die klinischen Symptome der adrenalen Hyperandrogenämie (Tabelle) und bei Männern auch das Wachstum der testikulären adrenalen Resttumoren (TART). Da diese Tumoren gutartig sind, müssen der mitbehandelnde Urologe und der Patient bezüglich der Grundkrankheit AGS gut informiert sein, da es sonst häufig zu unnötigen Hodenoperationen kommt.
Das vermännlichte äußere Genital kann operiert werden, damit Geschlechtsverkehr und Schwangerschaft möglich wird. Der optimale Zeitpunkt für eine Operation wird derzeit kontrovers diskutiert. Patienten mit nicht klassischem AGS bedürfen nur bei klinisch relevanten Symptomen einer Therapie. Im Kindesalter wird eher niedrig dosiert Cortisol in Tablettenform gegeben, während im Erwachsenenalter auch Antikonzeptiva ("Pille") eingesetzt werden, die die männlichen Hormone unterdrücken können.
Hilfreiche Internet-Adressen:
- AGS-Eltern- und Patienteninitiative: www.ags-initiative.de
- CARES Foundation: www.caresfoundation.org
Autoren:
PD Dr. H. Willenberg,
PD Dr. S. Hahner,
Prof. Dr. F. Beuschlein,
PD Dr. S. Diederich,
PD Dr. M. Fassnacht.,
PD Dr. M. Quinkler,
PD Dr. F. Riepe
für den Beirat der Sektion Nebennieren, Steroide und Hypertonus